


人源干细胞制剂作为细胞治疗领域的重要组成部分,在医学科技迅速发展的背景下,因其在疑难重症治疗中的潜力,受到全球政府和学术界的广泛关注。

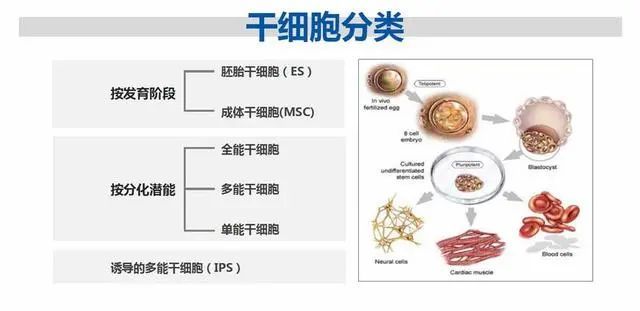
干细胞依据来源的差异,能够分为胚胎干细胞、成体干细胞以及诱导多能干细胞;依照分化潜能的不同,又可划分为全能干细胞、多能干细胞和单能干细胞。当下,临床所应用的干细胞制剂大多源自脐带、骨髓、脂肪组织等,属于成体干细胞的范畴。
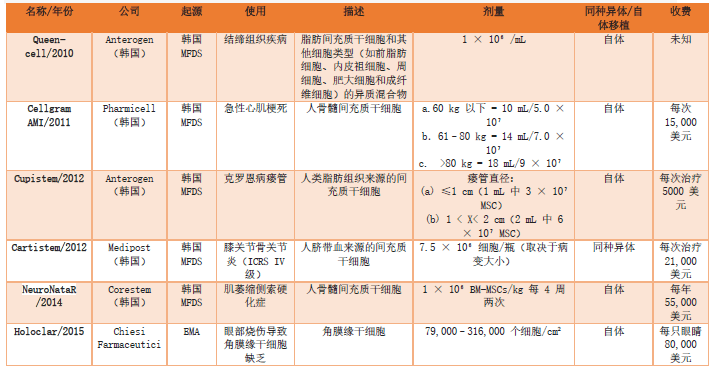
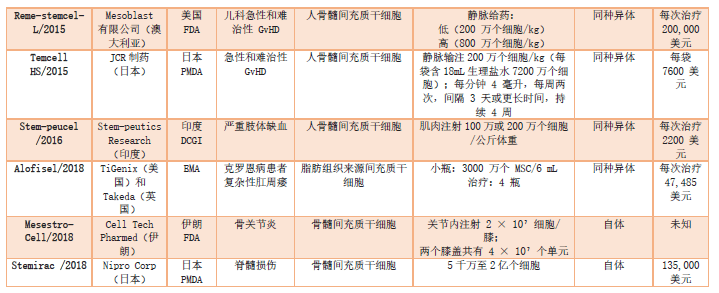
在国外,已有12款间充质干细胞制剂获批应用于临床治疗,其中韩国占据5款,欧盟有2款,日本为2款,美国、伊朗和印度各有1款。此外,还有诸多研究正处于临床试验阶段,其涉及的领域涵盖呼吸系统、骨骼系统、肝脏、肾脏、神经系统、免疫系统等多个方面。
在我国,干细胞研究已被纳入《“健康中国 2030” 规划纲要》与《“十四五” 生物经济发展规划》之中。




通过药品临床试验登记与信息公示平台进行查询能够发现,自2013年起直至现今,有关间充质干细胞的临床试验总计 45 项,其中 8 项已完成临床研究工作,37 项尚处于研究进程之中。
于细胞制剂临床应用发展过程中的微生物感染检测及相关规范
为了确保干细胞类产品在临床中的安全和规范使用,干细胞制剂需要经过制备、质量控制、临床前研究和临床试验等多个环节。在此过程中,微生物感染防控尤为关键,但传统微生物检测方法(如培养法)存在耗时长和灵敏度不足的问题,无法满足干细胞制剂快速放行的需求。新兴的宏基因组测序和质谱技术为微生物检测提供了更有效的解决方案。
1.干细胞临床应用中的微生物污染控制环节
干细胞临床应用涵盖原材料采集、制剂制备、放行检验、治疗等关键环节,各环节微生物污染控制方法及参考规范详见表1。


中国医药生物技术协会于2023年7月发布的《细胞治疗产品生产用原材料的质量管理规范》规定生产用原材料是指在细胞治疗产品生产过程中与产品直接接触但不进入终产品的材料,如培养基、添加因子、血清等。这些材料可能影响产品的安全性和有效性,因此需要采取措施防止病源微生物的引入。国家药典委员会在《中华人民共和国药典》中对这些材料的定义与此类似,并排除了生物制品生产的起始原材料。
国家药品监督管理局药品审评中心 2023 年 4 月发布的《人源干细胞产品药学研究与评价技术指导原则(试行)》对干细胞制剂生产用原材料定义范围较广,包含起始原材料及其他原材料多种类型,由此体现出干细胞制剂质量控制除起始材料无菌性外,还有更广泛的微生物污染防控要求。
前两者主要强调对相关原材料有严格无菌要求,像供应商要验证病毒去除工艺、确保动物源性原材料无病原体,人源性原材料按国家标准检测病毒且使用者要确认材料适用性,血清类材料参照药典标准检测,同时指出在实际操作中应借助技术进步加强新兴检测技术开发应用,来更好地满足干细胞制剂微生物控制需求。
1.2干细胞治疗产品制备过程中的微生物污染控制
干细胞制剂制备过程中的微生物污染控制包括对供者的筛选、采集过程的标准操作、接收/制备/储存/放行检验/运输等过程的无污染控制。为保证干细胞制剂在临床的安全使用,应严格控制每一个可能引入污染的环节:
(1) 在干细胞供者筛选和采集过程中,必须对供者的健康状况进行全面评估,重点检测传染性和遗传性疾病,检测项目包括但不限于人类免疫缺陷病毒、乙型肝炎病毒.丙型肝炎病毒等,确保供者无传染性疾病风险;同时,在采集过程中要严格执行无菌操作,确保干细胞制剂的安全性和有效性,防止疾病传播。
(2)干细胞接收及制备流程需确保每个环节操作无菌,并严格选择培养基、辅料、包装材料等物料,保障干细胞富集、分离、纯化、扩增等过程的安全性与质量,同时确保细胞系细胞库的建立和向功能性细胞的定向分化过程符合标准。
(3)干细胞制备过程必须严格遵循无菌、无病毒、无支原体及低内毒素原则,并遵循国家发布的相关法规和指导文件,如2015年8月,原国家卫生计生委、食品药品监管总局联合发布《干细胞制剂质量控制及临床前研究指导原则(试行)》及国家药品监督管理局药品审评中心于2023年4月发布的《人源干细胞产品药学研究与评价技术指导原则(试行)》,以规范干细胞制备过程,确保质量控制,防止微生物污染,保障干细胞产品的临床安全性。
1.3干细胞制剂放行检验过程中的微生物污染控制
《中华人民共和国药典》第四部要求药品和生物制品进行至少14天的微生物培养检测无菌性,但细胞类制品因效期短、剂量少,现有微生物检测方法难以满足需求。为此,随着微生物检测技术的发展,药典第四部引入了多种快速检测方法,如呼吸信号法等,能够将培养时间缩短至3-7天。2021年和2022年发布的相关指导原则和标准进一步提出了细胞类制品的快速微生物检测方法。然而,细胞制剂在临床试验中的活力问题仍未解决,需开发新的检测技术。
1.4干细胞临床治疗过程中的微生物污染控制及留样检验
干细胞临床干预中,通过采集感染疑似患者的标本进行微生物检测,若发现感染,则采取相应治疗。同时,干细胞制剂放行检测实验室对留样标本进行溯源检测,以评估感染微生物的来源,并长期保存用于检测致瘤基因等。此留样检验机制为干细胞制剂的安全使用提供保障,帮助监控治疗中的微生物污染风险。
2 干细胞制剂现行微生物检测方法
目前,微生物检测主要依赖培养法和基因检测法。培养法是金标准,但耗时较长,不适用于细胞类产品放行检验;基因检测法则快速灵敏,能满足时间要求,但无法区分微生物是否存活。
2.1培养法
药典第四部通则介绍了多种微生物检测方法,主要包括无菌检测、细菌内毒素检测和支原体检测。无菌检测方法(如直接接种法和薄膜过滤法)要求培养时间至少14天,而快速微生物检测法如呼吸信号法可缩短时间至7天,但仍难以满足细胞类制剂的放行需求。细菌内毒素检测采用鲎试剂法,并可通过凝胶法或光度法进行,若样品干扰严重需寻找新检测方法,如重组C因子法。支原体检测则需要28天(培养法)或约10天(指示细胞法),建议研发更快速有效的支原体检测方法以满足干细胞制剂的需求。
2.2 基因检测法
微生物基因检测方法(如PCR)具有快速、准确、高灵敏度的优势,适用于药品制剂中的微生物检测。根据药典的相关指导原则,基因检测可以用于干细胞制剂等微生物检测,但无法区分微生物是否存活。与传统培养法相比,基因检测提供了更高效的检测方式,尤其在紧迫的放行检验中,PCR等基因检测可以作为初步筛查,若检测为阴性,则无微生物感染;若为阳性,则需采用培养法进一步确认。

3 小结与展望
由于干细胞干预涉及伦理问题,各国在技术和制度上存在差异。欧洲、美国和日本的干细胞治疗技术较为完善,欧洲推荐使用常规培养法和快速检测技术(如PCR、流式细胞术等),美国则提出快速检测法并更新了标准,日本通过法规《再生医学安全法》和《药品和医疗器械法》保障干细胞治疗的发展。
我国药典指出,传统微生物培养法是微生物鉴定的金标准,但耗时较长。核酸检测虽能提供快速检测,但仅能鉴定特定微生物,且多次检测增加成本和工作量。目前,缺乏有效的同时检测干细胞制剂中多种微生物的方法。宏基因组测序和质谱分析技术在微生物检测中具有高通量、无需培养、可检测多种病原体等优点,但尚未广泛应用于干细胞制剂的微生物检测。建议未来将这些技术标准化并纳入干细胞制剂的放行检验,以提高检测效率和准确性。
参考文献
[1] 中国医药技术协会.《干细胞制剂制备质量管理自律规范(征求意见稿)》[S].20161025.
[2] 药品临床试验登记与信息公示平台. 检索词“间充质干细胞”[EB/0L].[20241204].http://www,chinadrugtrials.org.cn/clinicaltrials, searchlist. dhtml.
[3] 中国医药生物技术协会.《细胞治疗产品生产用原材料的质量管理规范》[S]. 202307
[4] 国家药典委员会.“生物制品通则”中“生物制品生产用原材料及辅料的质量控制”[M]//《中华人民共和国药典》。2020:3-8.
[5] 国家药品监督管理局药品审评中心,《人源干细胞产品药学研究与评价技术指导原则(试行)》[S].20230427.
编辑|LiYe.ZG
审核|Geng.ZG